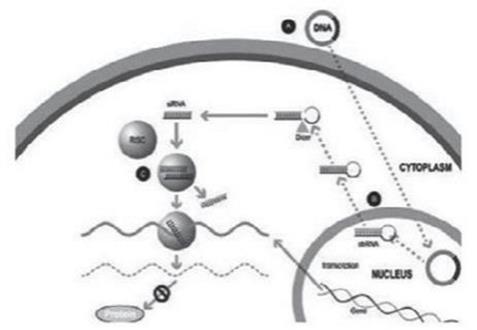
Figure 1
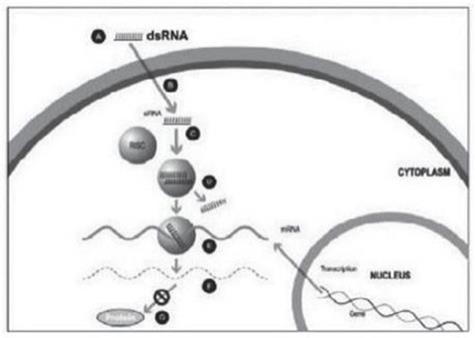
A small double stranded RNA, or dsRNA, molecule (A, Figure 1), comprising one strand known as the sense strand and another strand known as the antisense strand, which are complementary to each other, is synthesized in the laboratory. These small dsRNAs are called small interfering RNAs, or siRNAs. The sequence of the sense strand corresponds to a short region of the target gene mRNA. The siRNA is delivered to the target cell (B, Figure 1), where a group of enzymes, referred to as the RNA-Induced Silencing Complex, or RISC, process the siRNA (C, Figure 1), where one of the strands (usually the sense strand) is released (D, Figure 1). RISC uses the antisense strand to find the mRNA that has a complementary sequence (E, Figure 1) leading to the cleavage of the target mRNA (F, Figure 1). As a consequence, the output of the mRNA (protein production) does not occur (G, Figure 1). Several companies, including Alnylam Pharmaceuticals Inc. (“Alnylam”), utilize this approach in their RNAi product candidates.
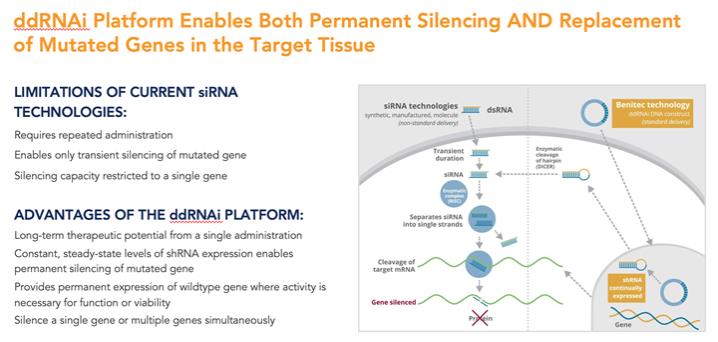
Importantly, many genetic disorders are not amenable to the traditional gene silencing approach outlined in Figure 1, as the diseased cells may produce a mixture of the wild type protein of interest and the disease-causing mutant variant of the protein, and the underlying genetic mutation may be too small to allow for selective targeting of the disease-causing variant of the protein through the use of siRNA-based approaches exclusively. In these cases, it is extraordinarily difficult to selectively silence the disease-causing protein without simultaneously silencing the wild type intracellular protein of interest whose presence is vital to the conduct of normal cellular functions.
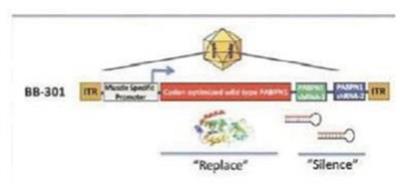
Our proprietary silence and replace technology utilizes the unique specificity and robust gene silencing capabilities of RNAi while overcoming many of the key limitations of siRNA-based approaches to disease management.
In the standard RNAi approach, double-stranded siRNA is produced synthetically and, subsequently, introduced into the target cell via chemical modification of the RNA or alternative methods of delivery. While efficacy has been demonstrated in several clinical indications through the use of this approach, siRNA-based approaches maintain a number of limitations, including:
| • | Clinical management requires repeat administration of the siRNA-based therapeutic agent for multiple cycles to maintain efficacy; |
23